Introduction
Sickle cell anemia (SCA) is a prevalent molecular disorder that has brought awareness to the hemoglobinopathies disease category and helped to encourage new treatment and screening techniques in largely affected areas. It is the most common sickle cell disease (SCD), accounting for approximately 83% of all newly diagnosed cases.1 In the Middle East and Sub-Saharan Africa, it is estimated that 300 000 new cases of SCD are documented a year in newborns.2 Due to migration, the incidence of SCA has spread beyond tropical regions in the world, which has sparked increased awareness and disease burden, and in 2006, led the World Health Organization to recognize SCA as a global public health problem. In the US, 1 of every 365 African American newborns is diagnosed with SCA.3 To address the growing prevalence of this disease, efforts in the field of stem cell research offer novel therapies that would allow for more direct treatment of the underlying disease etiology. This review aims to highlight and compare these new treatments and scientific advances with the current standard of care for SCA.
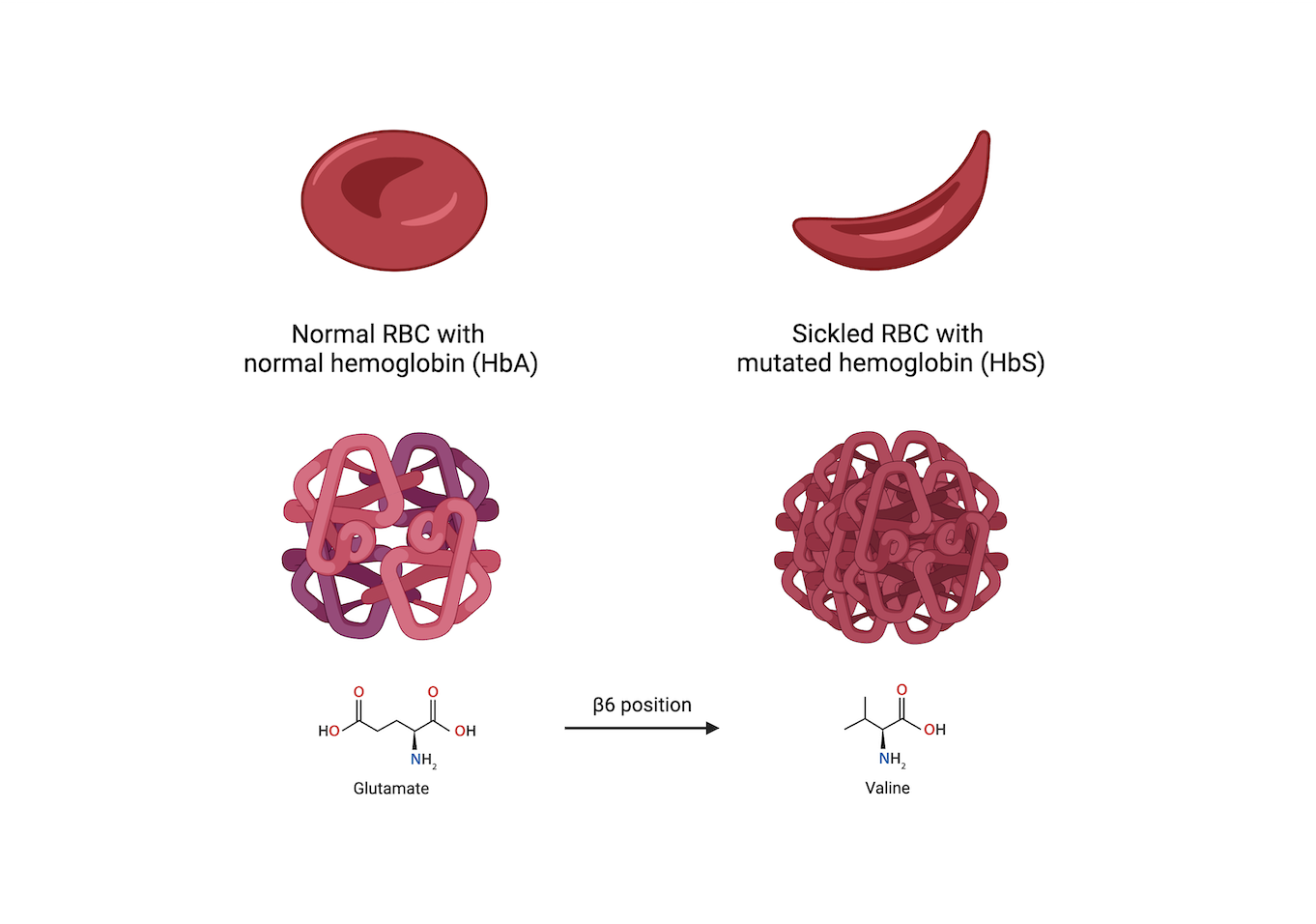
SCD refers to the category of diseases associated with abnormalities in hemoglobin (Hb) that lead to characteristic changes and behavior of red blood cells (RBCs). SCA is the most prevalent hemoglobinopathy among SCDs and is caused by a base pair alteration in the gene code for Hb, creating the sickled phenotype HbS.4 HbS polymerizes into abnormally long rigid chains under deoxygenated conditions in the vasculature, causing the surrounding RBCs to alter shape and assume a sickle-like appearance.5 This phenotype is observed whether the patient is homozygous recessive for HbS or heterozygous with the dominant form HbA.2 HbS differs from HbA by a single substitution of the amino acid glutamate to valine at the β6 position, affecting how individual Hb molecules interact within the body’s vasculature (Figure 1).5 These aberrant sickled RBCs exhibit different characteristics from healthy RBCs, which underlie most of the pathophysiological manifestations associated with SCA.
_with_normal_hemoglobin_(hba)_vs_sickled_rbc_wi.png)
This alteration in morphology reduces the deformability and lifespan of an RBC, while concurrently increasing its fragility and propensity to lyse in narrow vasculature.2 Sickled RBCs also exhibit increased adhesion molecules within the cell membrane, which prompt interactions and adherence to vascular endothelial cells. This modification in blood cell–endothelium interaction facilitates the entrapment of sickled cells in smaller vessels throughout the body, culminating in potentially harmful vaso-occlusive events as well as systemic anemia.6 These mechanisms are primarily responsible for the acute and chronic pain that many patients with SCA experience.
Vaso-occlusion can instigate a series of pathologies, including transient ischemia, stroke, and endothelial injury.2 Additionally, prolonged vascular tolerance of sickled RBCs leads to extensive cascades of inflammation and the potential activation of platelet and white blood cell mechanisms as a result of injury to the endothelium, ultimately leading to organ damage and dysfunction.2 Both the adhesion to endothelium and increased cellular fragility cause sickled RBCs to lyse significantly more than normal RBCs, further contributing to systemic anemia.6 New studies have also suggested that due to the altered deformability of sickled RBCs, system-wide blood flow can be affected and additionally contribute to tissue ischemia, endothelial injury, and the risk of aneurysm.7 Recent research in rheology and biophysics has added to the understanding of many SCA-related pathophysiologies. However, genetics and stem cell research have focused on aiding diagnosis and developing novel therapies to correct the point variant underlying the formation of sickled RBCs instead of solely addressing the symptoms of the disease.
Standard of Care and Pain Management
Treatments for SCA have traditionally targeted the management of symptoms and prophylactic interventions for SCA-related complications. Presently, conventional therapies include preventive treatments, chronic blood transfusions, and novel antisickling agents. The mainstay of current treatment is hydroxyurea, taken with folic acid supplementation to mitigate the risks of the drug.8 The primary therapeutic benefit of hydroxyurea in SCA comes from its antisickling properties, which translates to a 50% reduction in pain crises.8 The antisickling effect itself does not occur due to the direct use of hydroxyurea. Rather, hydroxyurea causes dose-related increases in fetal Hb (HbF), inhibiting in vitro sickling by preventing the polymerization of deoxygenated HbS.9 As such, hydroxyurea is only effective as a prophylactic therapy and not useful in the treatment of acute pain crises. However, this treatment comes with substantial risks and other adverse effects including, but not limited to, neutropenia, thrombocytopenia, myelosuppression, and erythrocytosis.8,9 Additionally, hydroxyurea is contraindicated in pregnancy because teratogenicity may be a risk with large doses.8 Another current treatment option is simple transfusion. This therapy is used for acute complications of SCA including, but not limited to, splenic sequestration, cardiovascular and cardiopulmonary dysfunction, and decreased oxygen delivery.8 Simple transfusion is contraindicated in uncomplicated pain crises, which the administration of analgesics may better treat.
Aside from continuous monitoring for the many potential complications that may arise in SCA, bacterial infection prevention via immunization and antibiotic prophylaxis is crucial to increasing positive patient outcomes.8 Specifically, affected patients have increased susceptibility to pathogens including Haemophilus influenzae and Salmonella, as well as to pneumococcal and meningococcal infections.8 Despite the use of preventive measures, infection may occur and has the potential to severely escalate, which most likely would require hospitalization and treatment with intravenous (IV) broad-spectrum antibiotics.8
The most common acute exacerbation of SCD manifests with vaso-occlusion, hemolysis, and tissue hypoxia, which may progress to ischemia and infarction of several organ systems.8 This intermittent condition is often termed pain crisis because it is accompanied by episodes of severe pain in the extremities. These pain crises are particularly distressing and may significantly decrease the quality of life (QoL) for patients. Clinicians must be diligent in their management of these crises through the use of analgesic agents such as opioids or nonsteroidal anti-inflammatory drugs. Continuous or bolus IV morphine administration has been demonstrated to be both effective and safe.8 Nonsteroidal anti-inflammatory drugs may also be used in addition to a lower dose of opioid analgesics to lessen the risk of addiction. Additionally, it is not recommended to use meperidine because adverse effects have been documented due to the accumulation of its neurotoxic metabolite, normeperidine.10 A thorough patient history should be taken before administration of any drug, paying particular attention to kidney failure, seizures, or history of acute chest syndrome as contraindications to common analgesic drugs.
Newer pharmaceutical developments, such as L-glutamine, crizanlizumab, and voxelotor, are promising alternative drug therapies for SCA, although their efficacies have yet to be fully established.8 Further research through randomized clinical trials should be done to determine the therapeutic benefits and potential adverse effects of these drugs and their comparison with conventional treatment with hydroxyurea. However, it is worthwhile to note that, like hydroxyurea, these drugs also target symptom management and pain crisis prevention and are not to be considered curative measures.
Hematopoietic Stem Cell Therapy for Sickle Cell Disease
The development of stem cell transplant may offer a long-term solution for SCD; however, multiple methods are currently under consideration. The most common method continues to be allogeneic stem cell transplant.11 Allogeneic stem cell transplant works by matching the human leukocyte antigen (HLA) alleles (HLA-A, HLA-B, HLA-C, and HLA-DRB1) between donor and recipient, with an identical sibling match offering the most significant success.11 However, an HLA-identical sibling is often challenging to find, so other transplant options, such as mismatched-HLA, haploidentical (HAPLO), and autologous transplants, are matters of current research.11 Although promising, the use of any donor besides an HLA-identical sibling increases the risk of mortality and graft failure.11 Due to the vast amount of polymorphisms present in the 8 HLA loci, many people cannot find an appropriate donor.12 Donor chimerism is the terminology used to describe the extent of replacement of the recipient’s bone marrow or other stem cell sources from the donor.13
If an appropriate donor is found, 2 avenues to explore with umbilical cord blood are to harvest either early-on or later-in-life donations through an adult donor.12 The ability to find an ideal donor that matches all 8 HLA alleles is highest in the White European population (76%) and decreases to 16% in the population of African descent.12 The probability of finding a suitable donor using cord blood (6/6 identical match) falls to even lower odds for both European and African populations (28% and 6%, respectively).12 The limited availability of HLA-identical siblings indicates the need for further research into autologous stem cell transplant.5,11
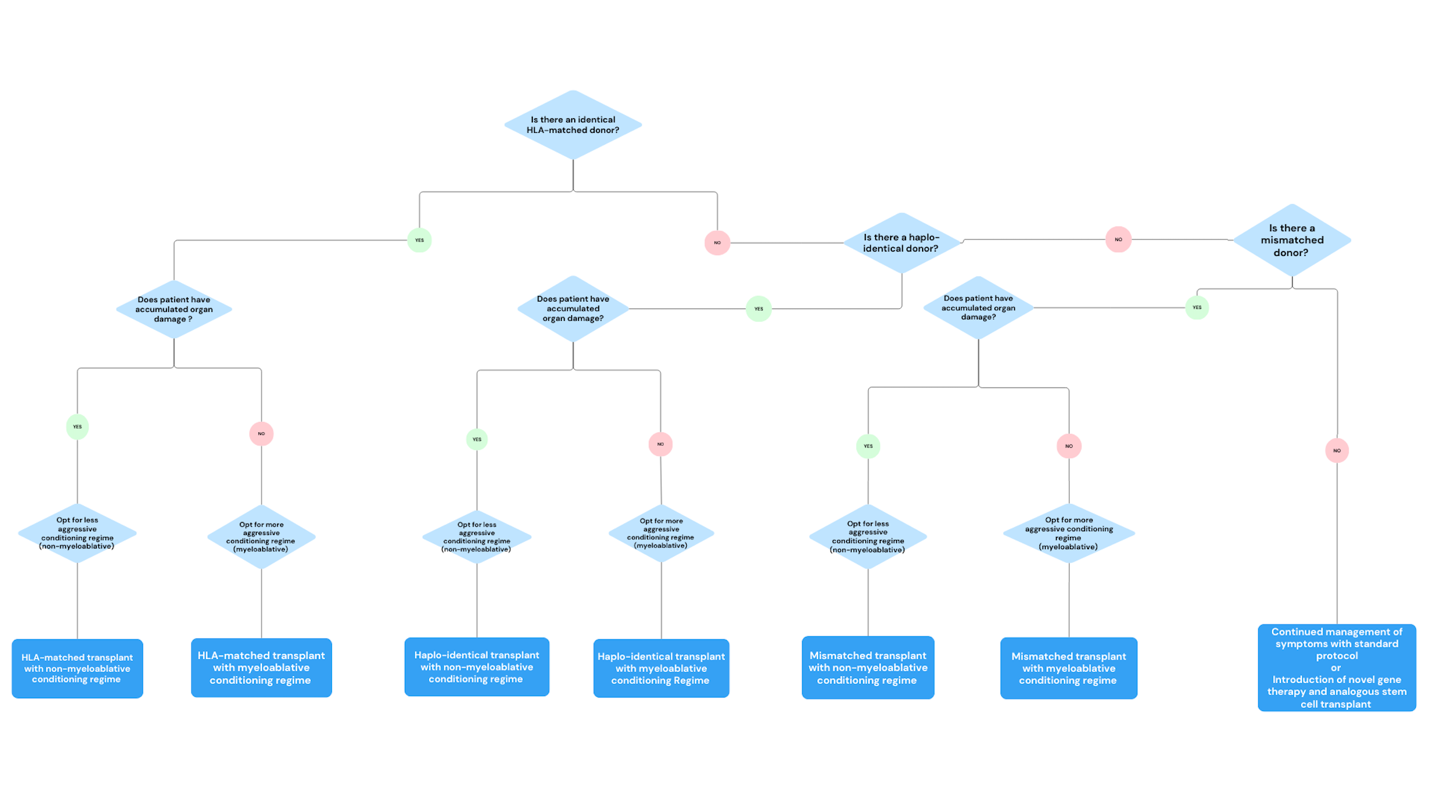
Along with multiple sources of transplant, there are differing conditioning regimens to prepare each patient for stem cell transplant. The most intense conditioning method is myeloablative, which uses chemotherapies such as busulfan (8 mg/kg by mouth or 6 mg/kg IV) or melphalan (150 mg/m2).11 In contrast, nonmyeloablative conditioning is considered less intense because it forgoes chemotherapy drugs in favor of total body irradiation at doses between 200 and 400 cGy.11 In the pediatric population, allogeneic stem cell transplant has shown excellent improvement in disease-free survival; however, there are less encouraging results in the adult population.11 As patients continue to live with SCD, the damage done to their organs accumulates.14 Some risk factors associated with transplants are the development of chronic or acute graft-versus-host disease (GVHD) and full rejection.11 While there is less risk of GVHD when using myeloablative conditioning, the organ damage accumulated from SCD makes it difficult for the patient to undergo aggressive conditioning.14 In light of these many factors, physicians must consider the patient’s condition in choosing the best method of transplant therapy for the patient (Figure 2).

Pediatric Studies Associated With Allogeneic Hematopoietic Stem Cell Therapy for SCD
Since the late 1980s, patients younger than age 30 years have undergone hematopoietic stem cell therapy (HSCT) to alleviate symptoms and cure SCA.15 The long-term outcomes of 234 patients with SCA (median age, 8.4 years [range, 2.2-28.9 years]) who received myeloablative conditioning and a matched-sibling stem cell transplant, were analyzed by a French research team.15 A 5-year event-free survival rate of 93.9%, which included incidences of rejection, graft failure, and death, was reported.15 Accounting for patients followed up since 2000, there was a 95% potential for SCA cure with matched-sibling HSCT.15 Evaluating the long-term outcomes, a more than 70% donor chimerism was found in 112 patients at their last recorded visit.15 As referenced in the above section, there is a small matched-sibling donor pool. Therefore, it is important to consider additional sources of stem cells to improve the QoL of patients with SCA, while ensuring patient safety.
A 2020 research letter detailed the findings in a prospective phase 2 multicenter trial consisting of 19 pediatric patients with SCD using a HAPLO stem cell transplant from a family member.16 Noting the median follow-up was 1409 days, the incidences of different acute GVHD stages and moderate and/or severe chronic GVHD were 6.2% and 6.7%, respectively.16 The study team also calculated the probability of overall survival at 1 year and 2 years post transplant being 90% and 84%, respectively.16 Following transplant, SCD-related symptoms were not reported in any recipients.16 The study team referenced a long-term follow-up study that aims to further evaluate chronic GVHD risks, overall QoL, and other variables.16 HAPLO stem cell transplant is an important area of study to increase the donor pool. Although there is still a risk of GVHD, increased research for pretransplant and posttransplant care will aid in decreasing this percentage.
Due to the significant risk of stroke in children with SCA, it is recommended that affected pediatric patients undergo annual screening using a transcranial Doppler flow study between ages 2 to 16 years.8 Chronic exchange transfusion therapy is one avenue of care that may help prevent cerebral thrombotic events. However, exchange transfusion is contraindicated for patients with an initial hemoglobin level less than 7 g/dL, although this may be remedied by red cell transfusion.8 To address the aforementioned contradiction, a nonrandomized controlled intervention study attempted to demonstrate whether HLA-matched sibling transplants in patients younger than 15 years lowered the risk of ischemic stroke in patients with SCD.17 In the trial, transcranial Doppler ultrasonography was used to measure the time-averaged mean of maximum velocities (TAMV), with higher velocities correlating with an increased risk of stroke.17 TAMV was recorded on initial visit and at the 1-year and 3-year follow-ups for children undergoing matched-sibling donor HSCT and the current standard of care.17 The study provided a promising alternative because the TAMV in the transplant group was significantly lower than in the standard group. Additionally, the number of participants in the transplant group at a normalized TAMV was greater than that of those in the transplant group.17 While this study demonstrated the efficacy of matched-sibling donor HSCT in reducing the risk of stroke, it should be noted that infertility, premature death, and transplant rejection are adverse events associated with matched-sibling donor HSCT.17 These associated adverse events are a common concern discussed throughout this article.
Adult Studies Associated With Allogeneic HSCT for SCD
To best address the toxic nature of myeloablative allogeneic HSCT, there have been efforts to explore nonmyeloablative allogeneic HSCT in adults. A nonmyeloablative conditioning regimen consisted of pretransplant alemtuzumab administered via gradual dosing and 300-cGy total body irradiation, and pretransplant and posttransplant sirolimus was used to treat 10 patients with severe SCD prior to receiving a sibling HLA-matched allogeneic HSCT.18 A median of 30 months of follow-up reported that all patients survived, but only 9 of 10 patients had successful engraftment.18 It is important to note that no patients experienced GVHD.18 Sirolimus was continued post transplant as a safety precaution given that all patients had less than 100% donor chimerism T cells.18 This research surrounding nonmyeloablative conditioning was a step forward, but there are still necessary steps to show efficacy. These include increasing patient enrollment in clinical trials and ensuring long-term follow-up with patients to assess the stability of donor chimerism as well as possible effects of the pretransplant and posttransplant regimen.
A follow-up study from Hsieh and colleagues13 included 20 additional patients, follow-ups on the original 10 patients, and an amended protocol. The amended protocol included a tapering of sirolimus in participants due to the stability of donor engraftment and the reduced incidence of adverse events attributed to long-term use of the drug.13 At a median of 3.4 years post transplant, 29 recipients survived and 26 recipients had engraftment.13 Full donor-type Hb was reported in 83% of recipients 1 year post transplant, and for the duration of the study follow-up, no patient experienced GVHD, including patients who met the guidelines to stop immunosuppression.13 The mean hospitalization rate analyzed decreased every year up to 3 years post transplant, and for patients taking long-term narcotics, the average usage was decreased by approximately 500 mg of IV morphine equivalent.13 The continued monitoring of the aforementioned 10 patients and increased participation provided further encouragement for the use of allogeneic HSCT with nonmyeloablative conditioning to cure SCD. A promising aspect of this continued research was a reduction in narcotics usage and a decline in hospitalization. As mentioned in the Standard of Care and Pain Management section, pain management for patients with SCD is an important aspect of holistic care. However, reducing the need for prolonged use of narcotics will help to mitigate the potential consequences of long-term reliance.
Saraf and colleagues14 validated the results of the above-mentioned nonmyeloablative allogeneic HSCT in a separate cohort of patients. There is potential that regimens that use nonmyeloablative conditions without chemotherapy may confer improvements in both QoL and disease progression, while limiting the risks associated with more aggressive forms of myeloablative conditioning.14 A group of 13 adults (17-40 years old) underwent HLA-identical matched-related donors with nonmyeloablative conditioning. The regimen consisted of permanent discontinuation of hydroxyurea a week before the transplant, a 300-cGy dose of total body irradiation 2 days before the transplant, and IV administration of alemtuzumab, which is an anti-CD52 monoclonal antibody.14 The trial demonstrated that this regimen normalized Hb concentrations in adults with advanced-staged SCD, while limiting the risk of GVHD associated with transplants occurring in adults undergoing myeloablative conditioning.14 While the sample size was small, this study demonstrated that there is potential benefit in using nonmyeloablative conditioning in patients who are older or have more advanced levels of organ damage.14 This regimen developed by the National Institutes of Health needs to demonstrate similar outcomes in a larger patient population but offers a potentially curative option for adults living with SCD.14 In addition to the validation provided, this study also assessed the QoL in patients undergoing nonmyeloablative allogeneic HSCT. Allogeneic HSCT recipients were asked to complete a short-form survey at baseline and 1year post transplant.14 At the 1-year follow-up, the recipients reported improved mental health and a dampening of symptoms associated with SCD.14 This finding shows the importance of HSCT in aggressive SCD, not only for its potential curative effects, but also for the ability to improve the day-to-day life of patients living with the clinical manifestations of SCD.
Autologous HSCT and Gene Therapy
HLA-matched donors have a reduced chance of graft rejection and lower mortality rate than recipients with an HLA-unmatched donor.11 As previously mentioned in this review, there are drawbacks to stem cell transplant for patients with SCD. With an appropriate HLA donor, disease-free survival and a potential cure can be achieved in most cases; however, less than 20% of patients with sickle cell have access to HLA-matched donors.19 To address the decreased availability of donor HLA-matched hematopoietic stem cells, there have been increased research efforts in autologous HSCT.
A single-center, open-label, pilot study explored the use of BCH-BB694 lentiviral vector–transduced CD34+ cells from 6 patients (age range, 7-25 years) with severe SCD for autologous HSCT.5 The expression of 𝛾-globin is downregulated via BCL11A and, subsequently, increased HbF creation is induced.5 BCH-BB694 lentiviral vector was designed with an embedded short hairpin RNA intended for BCL11A mRNA, which ultimately induces HbF production and decreases HbS as well.5 To mitigate adverse events, patients received transfusion to decrease HbS, stopped hydroxyurea therapy, and received myeloablative conditioning.5 During the study follow-up, all patients had posttransplant induction of HbF and there was a reduction in SCD-associated symptoms.5 Patients were able to opt into a 13-year follow-up study and the researchers noted that continued monitoring for potential diseases, including cancer, is necessary because such diagnoses can be associated with myeloablative conditioning or lentiviral vectors.5 Exploring autologous HSCT with genetic variations is a vital area of research, given the limited availability of HLA-matched donors, and creates more treatment opportunities. It is important to continue the long-term follow-up of patients to ensure the safety and efficacy of this new treatment. Given the potential toxicity and long-term effects of myeloablative conditioning, a potential area of future research would be pairing nonmyeloablative conditioning with autologous HSCT.
Another potential lentiviral vector–based autologous stem cell gene therapy has been researched. LentiGlobin is a gene therapy that uses BB305 lentiviral vector, which aids in the creation of HbAT87Q (antisickling hemoglobin) via a β-globin gene alteration.20 This lentiviral vector is transduced into hematopoietic stem and progenitor cells (HSPCs), allowing for autologous transplant in a phase 1/2 study with 35 participants with SCD in cohort C.20 Cohort C was developed for patients with SCD, following the analysis of smaller cohorts A and B and optimization of the protocol.20 Cohort C was divided into 2 groups: 25 patients matched the criteria for the transplant population with vaso-occlusive events group and 10 patients were in the transplant population group.20 Engraftment with LentiGlobin was observed in all 35 recipients and only 3 patients had nonserious adverse events associated with this gene therapy.20 For patients in the transplant population with vaso-occlusive events group, who were still being followed up, the study team noted that no severe vaso-occlusive events occurred following infusion of LentiGlobin.20 After 37.6 months, there were no reports of hematologic cancer in patients who received LentiGlobin.20 The authors acknowledged the need for more participants in the clinical trial and longer follow-up periods to assess the full safety and efficacy of LentiGlobin.20 Research of autologous HSPCs with gene therapy is a promising avenue but still necessitates more studies to determine the benefits and safety.
Because many SCDs, such as SCA, come from variants in the adult Hb gene, genetic modifications, such as CRISPR, should be discussed as a potential future therapy.21 In a genetic condition called hereditary persistence of fetal hemoglobin, both the fetal and adult forms of Hb are present.22 This benign condition allows patients with SCD to overcome the HbS by producing sufficient HbF, leading to fewer clinical manifestations of SCD.22 With this in mind, a single study using CRISPR technology focused on creating an alanine to glycine point variant of 113 base pairs upstream of the 𝛾-globin transcription start site.21 This study demonstrated that a single point variant was enough to induce the production of HbF by creating a novel binding site for GATA1 without disrupting the major repressors of HbF, such as BCL11A.21 However, in another study, CRISPR was used to create variants in the -98 and +68 promoter regions of the adult HBB gene of CD34+ HSPCS. This variant caused the production of HbF to increase in the CD34+ erythroblast.22 Both of these studies proved effective in inducing HbF through different mechanisms, thus indicating a promising future for using CRISPR in SCDs; however, due to the variation of mechanisms, more research is needed to find the best gene target.
Genetic variations in unrelated stem cell sources could be another area to expand the stem cell donor pool. A pediatric study explored the use of unrelated cord blood (UCB) and omidubicel, a nicotinamide-based UCB product, with a total of 16 participants (median age, 13 years) separated into 2 cohorts: 13 patients received unmanipulated UCB grafts with omidubicel and 3 patients received only omidubicel.23 For the cohort that received unmanipulated UCB grafts with omidubicel, long-term engraftment was attained with omidubicel in 2 patients and 10 patients with unmanipulated UCB.23 This cohort also had posttransplant infection occur in 11 patients and acute grade 2-4 GVHD in 10 patients.23 Chronic GVHD was observed at 6 months post transplant (15%) and 1 year post transplant (46%).23 Two patients died in this cohort: 1 of posttransplant infection and 1 of liver GVHD.23 Of the 11 surviving patients, there were no reported SCD symptoms or related events, and all were able to stop immunosuppression medication following resolution of GVHD.23 Of the 3 patients who received only omidubicel, 2 experienced GVHD and 1 experienced graft failure and needed to be regrafted.23 One year post transplant, 2 of the 3 patients had 100% donor chimerism and were able to stop immunosuppression.23 Using unmanipulated UCB and omidubicel has shown encouraging data, but still needs long-term follow-up, especially given the high incidence of posttransplant infection and GVHD.
Conclusions
Hemoglobinopathies are a class of molecular disorders that are challenging to treat and have prompted many avenues of symptomatic treatment as the current standard of care. The advent of stem cells has become imperative in the pursuit of better treatment options because these novel therapies provide a possible cure for SCA. As previously discussed, an HLA-identical sibling match is ideal for HSCT to mitigate risks to the recipient. Unfortunately, the difficulty in finding such a match necessitates other avenues for transplant such as HLA-mismatched, HAPLO, and autologous transplant with genetic variations. Any of these therapies require conditioning to prepare patients for transplant. Myeloablative conditioning, in particular, is toxic and uses chemotherapeutic agents. The next best option after an HLA-identical match may be a nonmyeloablative allogeneic HSCT, which will require increased research. Autologous transplant with genetic variations could provide a complete match of all 8 HLA alleles because the recipient is also the donor. Combining this method with nonmyeloablative conditioning may help provide a cure for which patients may achieve a state of disease-free survival, without many of the risks associated with current stem cell therapy or the inability to find a suitable donor.
Currently, the shortage of donor matches coupled with an increased risk of organ damage and failure with age complicates the HSCT transplant decision process.11 As such, this complex decision requires a balance between patient autonomy, ethical consideration, physician guidance, and family input.19 Particularly among the pediatric population, patient autonomy must be closely considered while working with family and other caregivers to respect all wishes when making medically appropriate treatment decisions. Additionally, it is crucial to consider the future fertility of many younger patients who undergo stem cell transplant. Myeloablative conditioning uses chemotherapy drugs such as busulfan, which may reduce ovarian function and may lead to infertility in both sexes.15
Stem cell transplant will positively affect the prognosis of many patients globally. However, many of these treatments require a high level of technological sophistication and resources that are not readily available in many regions affected by SCDs. Barriers to care, such as housing, transportation, financial stability, medical resources, and access to basic screening among vulnerable populations, have to be concurrently addressed to allow novel therapies to reach the populations most in need.3 Much of the research involving stem cells and genetic therapy is still ongoing. While many of these therapies could improve outcomes significantly for patients with SCA, ethical and resource pitfalls, optimization of treatments, and their proliferation to worldwide populations are primary concerns. As the foundational principles of these therapies are solidified, further research comparing the efficacy and safety of treatment options should be pursued to determine the new standard of best clinical practice and disease protocols.
Conflict of Interest
All authors have no conflicts to disclose.