Introduction
Cystic fibrosis (CF) is a multiorgan disease affecting the exocrine mucous glands of the lungs, liver, pancreas, and intestines.1 Affecting 1 in every 4000 neonates in the United States, CF continues to pose a significant disease burden.2 The disease is inherited in an autosomal recessive manner and results from a mutation on chromosome 7 in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.2 Up to 90% of patients with CF have a mutation in the form of a Phe508 deletion, resulting in an amino acid, phenylalanine, being deleted from the CFTR protein. However, numerous other CFTR mutations have been identified as causes of CF.3 The CFTR gene encodes an ion channel responsible for transporting chloride and bicarbonate across epithelial cell membranes. Aberrant ion channels along the lumen of respiratory tracts cause significantly reduced ion and water secretion into lumen mucus. This results in excessively thick and stagnant mucus, with a predilection to block the airways and acts as a nidus for infective organisms. Mutations in the lungs are phenotypically expressed as decreased airway clearance, increased airway mucus and subsequent bacterial infection, and lung damage.1 Although the disease impacts many organs, the respiratory manifestations of the disease are often the focus of therapies because targeting the lungs can significantly improve quality of life and decrease mortality in individuals with CF.4
The current management of CF consists of treatments targeted toward symptomatic improvements. However, a curative treatment targeting the underlying pathophysiology of the disease has yet to be clinically realized. This review outlines 3 main curative approaches at the forefront of CF research: stem cell–based therapies, gene therapies, and small molecule treatments. Small molecule treatments have demonstrated sufficient efficacy in human trials.1 Ninety percent of the disease-causing variants are responsive to current small molecule treatments.4 Stem cell–based treatments, which include mesenchymal stromal cell (MSC) therapy and induced pluripotent stem cell (iPSC) therapy, as well as gene therapies could potentially be effective in all the disease-causing variants. However, these treatment strategies are still in their infancy, and their safety and feasibility have yet to be determined.
Stem Cell Therapies
Mesenchymal Stromal Cell Therapy
MSCs are stem cells derived from the bone marrow and found in the umbilical cord and adipose tissue.5 Due to their dynamic anti-inflammatory properties, MSC therapies could potentially treat lung inflammation in patients with CF.5 Their anti-inflammatory properties are dynamic in that the environment that the MSCs are exposed to can alter the level of MSC cytokine expression.6 The ability for MSCs to produce anti-inflammatory signals in response to their environment was demonstrated in a study that involved the introduction of MSCs to bronchoalveolar lavage fluid (BALF) from patients with CF and acute exacerbations or coexisting acute respiratory distress syndrome (ARDS).6 As a control, patients with CF but without acute exacerbations or coexisting ARDS had their BALF extracted and exposed to MSCs.6 Notably, the MSCs exposed to patients with CF exacerbations or ARDS had an increase in the production of IL-6, a proinflammatory cytokine that potentially exacerbates neutrophil-induced inflammation and lung damage.6 In contrast, MSCs exposed to BALF from patients without CF exacerbations or ARDS exhibited significantly reduced IL-6 levels.6 This understanding of the environmental dynamics of MSCs was an integral step towards conditioning the MSCs as therapeutic agents for CF and other diseases.6 Thus, it can be inferred that MSCs will produce anti-inflammatory effects and favorable outcomes in patients with CF and lower levels of baseline inflammation.6
Unfortunately, the translation of in vitro MSC studies to in vivo applications is not straightforward. Notably, the ability of MSCs to function in the lungs of a patient with CF is compromised when the respiratory environment possesses pathogenic microbial activity.7 Therefore, researchers recommend that the lung environments of potential patients are assessed before using MSCs as a therapeutic agent.7 Being that CF is commonly associated with a large amount of inflammation and mucous secretions in the airways that create an opportune environment for fungi and bacteria, MSC treatment may have decreased efficacy for those with more infectious symptoms.7 A notable study tested this hypothesis by using similar techniques of introducing MSCs to BALF but instead with patients who had CF and an Aspergillus infection.7 Aspergillus produces a toxin known as gliotoxin, which was found to increase MSC death via a mitochondrial damage pathway.7 This study suggested that while introducing MSCs to patients with CF may be therapeutic, the treatment effect is also limited by the inflammation inductive microorganisms present in the respiratory system, because they can impede the regeneration of lung epithelial cells.7
The current research of delivering MSCs as a reliable therapeutic agent for CF remains in consideration for future practices. While MSCs can provide anti-inflammatory effects, the delivery mechanism for this type of treatment for patients with CF is not fully characterized or understood.5 Furthermore, the change in protein profile under stress-induced or nonstress-induced environments brings both advantages and disadvantages in a clinical setting. Its advantages are that the MSCs can be manipulated to provide a therapeutic effect to patients with CF, but the concern lies in whether this therapeutic effect can be maintained.6 Additionally, if an infection is present, it may also complicate the anti-inflammatory response by MSCs.7
The anti-inflammatory response by MSCs is mediated by the release of their extracellular vesicles, otherwise known as their secretomes. To circumvent the challenges of using MSCs entirely, researchers investigated whether extracellular vesicles could be used directly as a potential therapeutic.8 The extracellular vesicles were found to decrease IL-1B and IL-6 but not IL-8 levels.8 Thus, researchers concluded that the extracellular vesicles secreted by MSCs were the source of anti-inflammatory effects and could potentially be used directly instead of full MSC transplantation in the lungs.8 This finding emphasized the constantly changing landscape of MSC research, indicating the recency of the data on treatment and the refinement needed before gaining clinical utility as a curative agent for CF.
Induced Pluripotent Stem Cell Therapy
The development of iPSC therapy for the treatment of CF holds great potential. iPSCs can be derived from fully differentiated somatic cells, such as skin or blood cells, which are then reprogrammed to an undifferentiated state.9 Treatment with specific growth factors and signaling molecules can guide subsequent iPSC differentiation into specific epithelial progenitors and ultimately fully differentiated tissue-specific epithelial cells.9–13
Like human embryonic stem cells (ESCs), iPSCs have the capacity for self-renewal and in vitro differentiation into cell lineages of all 3 embryonic germ layers.9 Because iPSCs can be generated from somatic cell types, bypassing the need to destroy human embryos, iPSCs provide an ethical alternative to the use of human ESCs. Moreover, because these cells can be patient-specific, the risk of immunogenic transplant rejection and the need for immunosuppression therapy, in theory, could potentially be eliminated, though research on this treatment modality is still in its infancy.
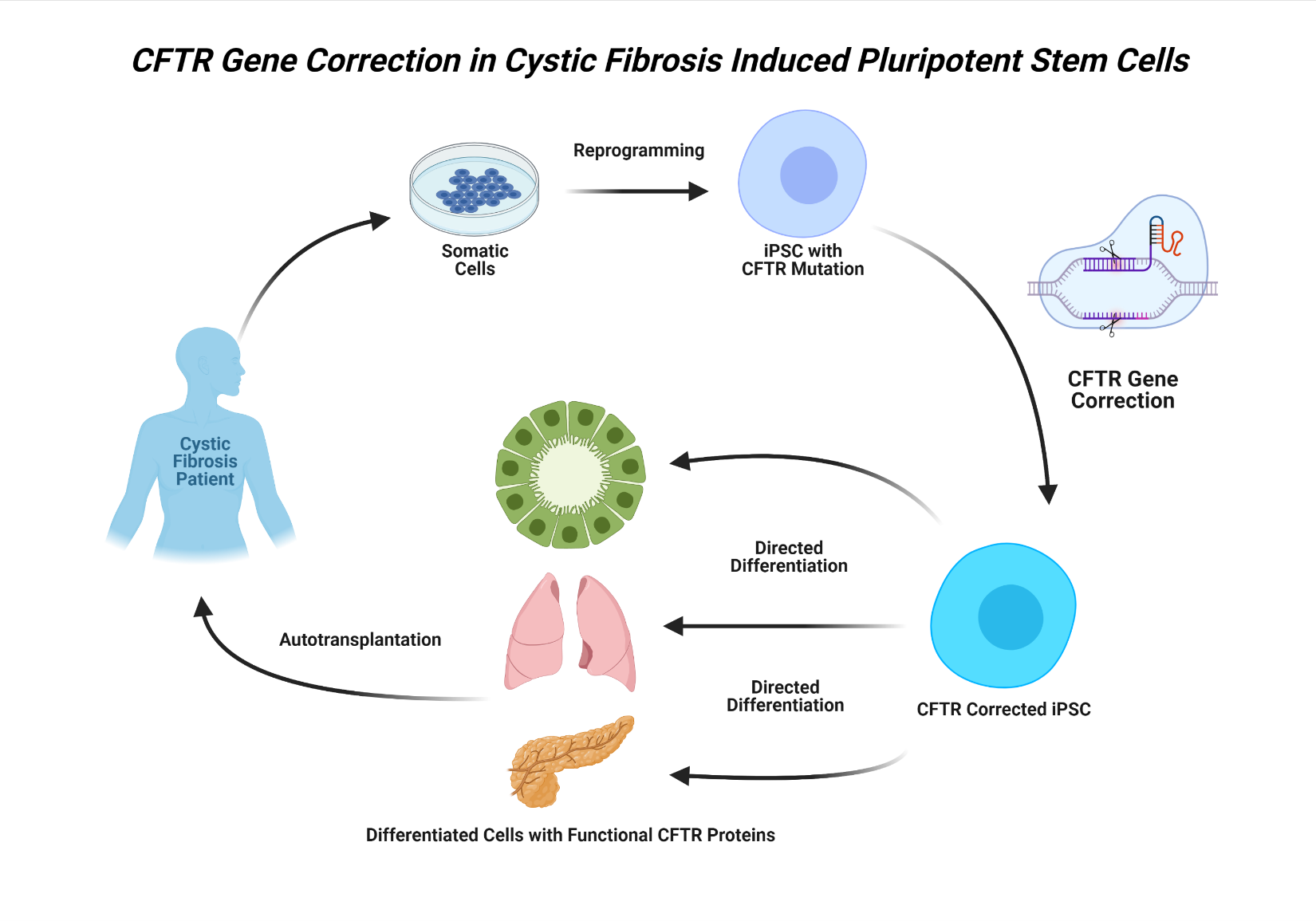
iPSC therapy in the context of CF would involve using somatic cells obtained from an affected individual to generate populations of patient-specific iPSCs.10 The iPSCs would then be directed to differentiate into tissue-specific progenitor cells in vitro and, after mutation correction using genome-editing technology and further cell differentiation, reintroduced into the donor.14 In theory, this treatment would essentially replace the diseased or defective tissue with iPSC-derived epithelial cells expressing functional wild-type CFTR proteins, restoring full tissue function (Figure 1).
_transmembrane_conductance_regulator_gene_correction_in_induced_plurip.png)
The first hurdle in developing pluripotent cell–based therapies to treat CF is developing a replicable and reliable method of generating large populations of progenitor cells capable of differentiation into tissue-specific epithelial cell types. Researchers have demonstrated success in generating patient-specific functional airway epithelial cells from pluripotent stem cells, such as ESCs and iPSCs, using signaling pathways similar to those involved in in vivo embryologic lung development.10,11 Using a carefully timed sequence of growth factors and signaling molecules mimicking the stages of embryologic development, Mou et al10 successfully produced iPSC-derived lung and airway progenitors capable of generating respiratory epithelium when subcutaneously transplanted into immunodeficient mice.
Although the primary focus of iPSC research has been on generating lung epithelium, the potential use of iPSCs for the regeneration of other organ tissues affected by CF is also being investigated. For example, researchers have generated fully differentiated cholangiocytes, epithelial cells lining bile ducts, from iPSCs obtained from patients with CF.12 Moreover, Simsek and colleagues13 demonstrated successful differentiation of wild-type and CF iPSCs into pancreatic ductal epithelial cells. Together, these findings suggest that, in addition to lung tissue, iPSC-based therapies may hold potential for the restoration of CFTR function in other tissues damaged by CF as well.
The true potential of iPSCs in the treatment and cure of CF centers around their use in combination with gene-editing technology, discussed in later sections of this review, to correct the mutations responsible for the defective CFTR protein. Using CRISPR/Cas9 gene-editing systems, researchers have demonstrated successful correction of the F508 CFTR gene deletion in CF iPSCs, yielding fully differentiated lung epithelial cells expressing functional CFTR proteins.15 Crane et al14 were able to correct the same mutation in CF iPSCs using zinc-finger nucleases (ZFNs) with similar results. While these findings are promising, the F508 deletion is one of many mutations responsible for the development of CF. Therefore, more research is needed to assess whether the efficacy of these methods would extend to other genotypes.
While pluripotent stem cell therapies for CF hold potential, many questions regarding their safety and clinical application remain to be answered. In addition to the safety concerns associated with genomic editing, the inherent potential for self-renewal and pluripotency of iPSCs raise concerns about potential tumorigenicity associated with iPSC-based therapies. Furthermore, the process by which gene-corrected iPSC-derived tissue will be transplanted back into the patient remains to be elucidated. Extensive research is needed before iPSC therapies for CF are prepared for clinical trials.
Gene Therapy
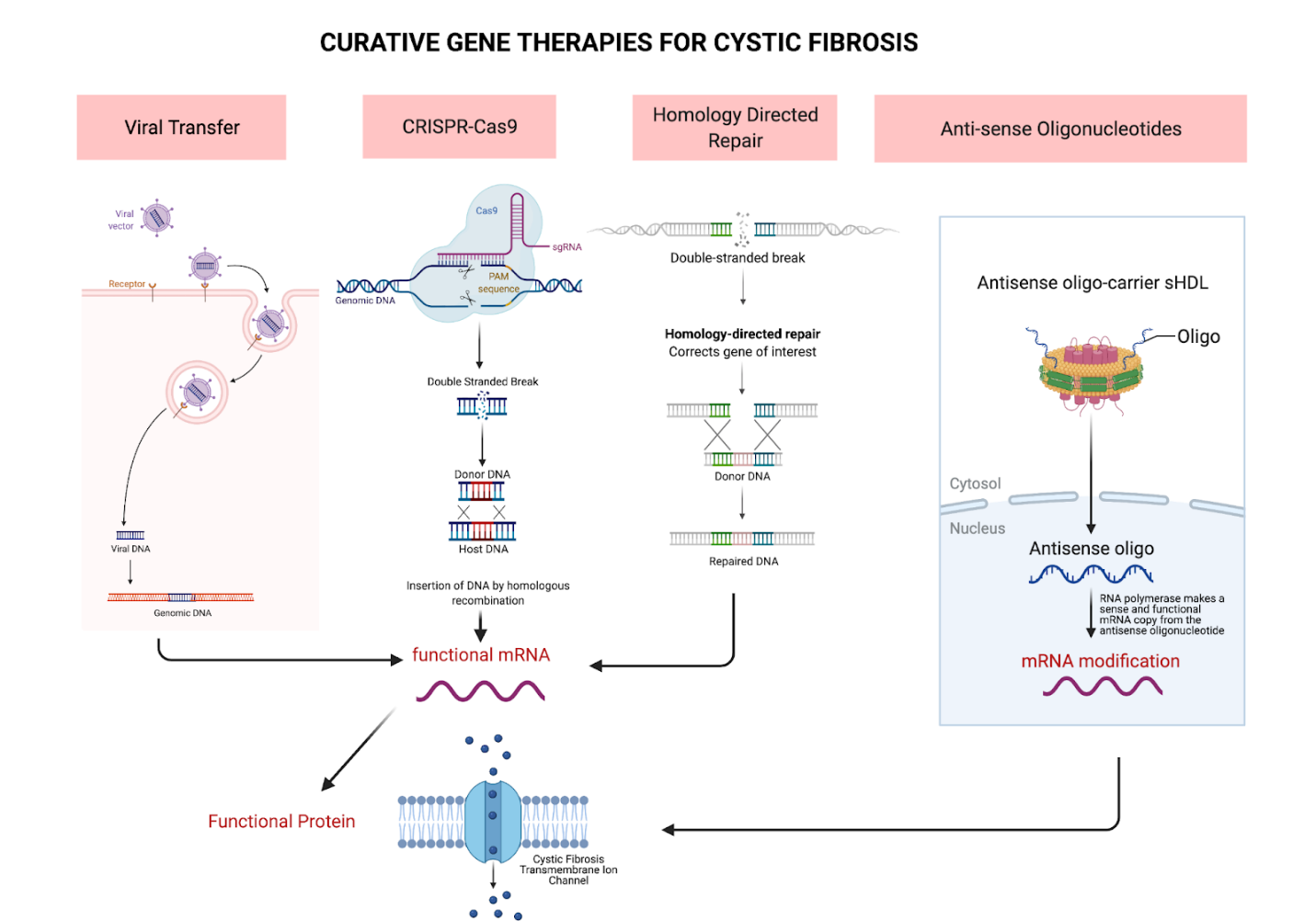
Advances in gene therapy provide promise towards finding a cure for CF. Gene therapy presents the opportunity to correct the underlying genetic error. Unlike CFTR modulators, a form of small molecule therapy, gene therapy has the potential to become a curative treatment in all phenotypic presentations of CF.1 However, there are many barriers to gene delivery that have yet to be overcome to make gene therapy a staple in CF treatment. Namely, pulmonary gene therapy has lagged in clinical development due to the numerous intra- and extracellular barriers. Although, inhaled gene transfer agents are able to achieve high drug levels in the airways without significant systemic adverse effects, barriers such as the sputum layer coating the airways of patients with CF impede their efficacy.1,16,17 Fortunately, the advent of drug delivery systems, such as the self-emulsifying drug delivery system, can help gene transfer agents achieve higher mucus permeability.18 Furthermore, effective treatment is predicated by the delivery mechanism’s ability to permanently alter progenitor epithelial cells of the lungs or continually deliver the genes to perpetually replaced terminally differentiated cells.1,19 Therefore, only a few methods of gene therapy have achieved clinically significant outcomes. The various gene therapy mechanisms are depicted in Figure 2. The research into these contemporary methods is still in its nascent stages, so the potential utility in these mechanisms for curing CF is yet to be fully understood or evaluated.

Viral Gene Transfer Mechanisms
Given the natural tropism of viruses to the respiratory tract, viruses were extensively studied as promising vectors for CFTR gene transfer. To apply this treatment clinically, the viral vector must be capable of efficient gene transfer and sufficient penetration of the enhanced mucus layer in CF fibrosis.20 However, despite numerous studies with different viruses, no viral vector has demonstrated all the requirements needed for clinical use.1 The location of receptors on the basolateral surface of the respiratory epithelium and the limitations resulting from preexisting or induced immune responses have resulted in poor viral vector gene transfer efficacy.1,21
Many viruses have been investigated as potential vectors for CF gene therapy. Among the first studied were the adenoviral vectors, which were shown to be unsuitable for gene transfer due to the lack of apical coxsackie-adenovirus receptors on human respiratory epithelium.21 Adeno-associated viral vectors were also investigated, only to show similarly unremarkable results when used to treat the maxillary sinuses of patients with CF due to their limited carrying capacity.22 Lentiviral vectors have the most promise as a CF treatment because they can transfect dividing and nondividing cells and have demonstrated a longer-term gene expression in their targeted cells.1 However, they carry the potential risk of insertional mutagenesis, and the research for this vector on CF specifically has yet to be conducted.1
In summary, viral vectors have yet to show considerable efficacy to be considered for CF treatment. Although lentiviruses have the most potential as an effective delivery mechanism, their use in CF research has not progressed to clinical trials, underlining the unsuitability of current viral mechanisms for CF gene therapy.
Nonviral Gene Transfer Mechanisms
Unlike their viral counterparts, nonviral vectors are not limited by packaging capacity or immune response.1 They include naked DNA, cationic liposomes, and DNA nanoparticles or polymers.1 A phase 1/2a study conducted by Alton et al23 investigating liposome-mediated gene therapy for patients with CF depicted a significant treatment effect and stabilization of lung function for patients who received the therapy.1 However, the investigators did not proceed to phase 3 studies due to the demonstrated variability in the treatment effects.1 Although these vectors have data showing their potential efficacy in CF treatment, they cannot transfect the apical surface of airway epithelial cells efficiently and are limited in their ability to traverse the cytoplasm to reach the nucleus.1
Homology-Directed Repair and CRISPR
Therapies using homology-directed repair (HDR) use the natural HDR process of the cell to correct the defective CFTR gene.24 HDR involves creating a double-stranded break on 2 ends of a target site and replacing the defective gene with a homologous and endogenous wild-type sequence.24 The most significant caveat of this method is that HDR occurs rarely, only 1 in 106 times, making it extremely difficult to experiment with and an ineffective treatment.24
Difficulties in the use of the viral and nonviral delivery mechanisms spurred the development of target integration using nucleases to provide a more pointed approach in correcting the defective CFTR gene locus. As opposed to therapy using retroviral vectors, programmable nucleases can target DNA sequences at specific cleavage sites.25 Targeted editing tools, such as ZFN, TALEN, and most commonly CRISPR/Cas9, are being studied for potential use in CF treatment.24
Whereas the previous mechanisms involved the delivery of functional genes, gene editing can correct the underlying genetic defect at its original locus. The ability to fix the core defect has the advantages of maintaining the endogenous promoter, allowing for sustained gene expression and natural regulation, and bypassing the risks of insertional mutations associated with foreign DNA.1 CRISPR/Cas9 is regarded as the best in genomic editing because its efficiency and simplicity far surpass that of ZFNs or transcription activatorlike effector nucleases.1 Studies have explored the potential of synthesizing stem cells with CRISPR/Cas9 gene editing in curing CF. For instance, Schwank et al26 found that CRISPR/Cas9 was able to correct the genetic defect at the CFTR locus in adult stem cells and provided lasting genetic and functional correction to the resulting clonally expanded organoids However, the research only suggests the plausibility of inserted CRISPR/Cas9-corrected stem cells into the lungs of patients with CF; in reality, there are no current trials affirming the viability or functionality of these cells in humans. Although the promise of a permanent fix is tempting, the usage of gene-editing mechanisms as a cure for CF requires much more investigation before it can be tested in human trials.
Antisense Oligonucleotide Therapies
Messenger RNA (mRNA) is another prime target that can be used in the treatment of CF. Antisense oligonucleotides (ASOs) are used to target gene regions with incorrect splicing instructions, modulating gene expression at the mRNA level, resulting in a functional protein.2,4 In cystic fibrosis, ASOs can be used to target the splicing mutation 3849 + 10 kb C T. This splice mutation causes the insertion of 84 new nucleotides and a subsequent nonsense mutation that culminates in a truncated CFTR protein.4 Studies of murine models demonstrated functional protein production after the administration of ASOs.4,27 ASOs can also be used to insert missing bases in the defective CFTR gene locus.4 Therapy based on mRNA is complicated by its transient nature; CFTR restoration through these methods is temporary, and treatment must be administered consistently for the entire length of the patient’s lifetime for the potential of consistent results.2
Small Molecule Therapy
There have been many pharmacologic interventions and innovations for the treatment of CF, notably CFTR modulators, which target the cause of the disease, misfolded CFTR proteins, as opposed to merely treating the symptoms.28 The first of these drugs to be approved by the Food and Drug Administration (FDA), ivacaftor, demonstrated a considerable improvement in lung function over placebo for patients with the G551D-CFTR missense mutation, a class III mutation found in approximately 4% to 6% of patients with CF.1,29 Ivacaftor allows the ion channel protein to open in the cell membrane.28 Although this drug was later approved for additional mutations as a monotherapy, ivacaftor’s limited therapeutic applicability to the broader CF population demonstrates the current issue with small molecule treatment: a lack of scope.1 Therefore, although these treatments are currently available for clinical use and have demonstratable treatment efficacies, their usages, especially as monotherapies, are limited to small subsectors of patients with CF with the specific mutations covered by the therapy.1
With the numerous possible mutations in the CFTR locus, small molecule treatments can be used with one another to maximize efficacy. This additive benefit was shown with the FDA approval of Orkambi (Vertex Pharmaceuticals), a combination of ivacaftor and lumacaftor, in 2015.1 By adding the CFTR corrector lumacaftor, which increases the number of the functional CFTR at the cell surface, the treatable population with small molecule therapy was significantly increased to cover most patients with CF.1 However, the combination of the 2 has been questioned for their clinical efficacy. The potentially antagonistic drug-drug interaction may explain the only modest improvements in lung function, which were at times eclipsed by the adverse effects for patients.1 A newer combination with tezacaftor, a next-generation CFTR corrector similar to lumacaftor, was also FDA approved for CF treatment and is expected to have a better benefit-to-risk profile than Orkambi for a larger population of patients with CF.1 The ivacaftor and tezacaftor combination has not yet had any reports of the same issues faced by Orkambi, showing promise for improved patient treatment outcomes with continued therapy.1
Another more recent combination drug, Trikafta (Vertex Pharmaceuticals), is a combination of ivacaftor, tezacaftor, and elexacaftor.3 All 3 compounds in this combination drug work on various components of the CFTR protein to stabilize it. Trikafta is approved for anyone carrying 1 copy of the F508 deletion, even if their second allele holds a different mutation.3
Further exploration into other additive therapies is currently underway, with many other CFTR-focused drugs currently in clinical trials.1 New combinations of CFTR stabilizers, potentiators, and correctors are currently being tested by pharmaceutical companies for greater efficacy across larger proportions of the CF population and improved safety profiles, including VX-445 and VX-659.1 Unlike the other categories of therapies previously mentioned, small molecule treatment is the only one with current clinical usage. The greatest challenge to its use, however, remains the applicability to the entire CF population. Outside of the patients with CF who have current FDA-approved treatments, ie, patients with the F508del, there are other patients who do not benefit from small molecule treatments due to the rarity of their mutations.1
Discussion
The management of CF has improved significantly since FDA approval of the first CFTR modulator, ivacaftor. Although small molecule treatments are not currently approved for the entire CF population, they were the first to provide treatment for the disease that extended beyond mere symptomatic relief.1 Through the combination of various CFTR modulators, new drugs such as Orkambi and Trikafta represent the current trajectory of the pharmacologic treatment of patients with CF.3,28 However, these drugs still have modest efficacy and do not have the same curative potential as the stem cell–based or gene therapies presented.1
The combination of contemporary stem cell research and gene-editing mechanisms provides hope for an eventual cure for CF, but none of the drugs are currently in a clinically relevant state. MSCs can alter their protein expression behavior with respect to changing environments of human lung tissue, but the anti-inflammatory effects of MSC transplantation may be hindered by patients with CF because of these stress-induced environments.7 From a clinical perspective, assessing practical candidates may pose an issue due to the dynamicity of a patient’s disease state.7 Further, obtaining BALF from a patient could be detrimental in relation to time consumption, resource availability, and patient satisfaction. While EVs secreted by the MSCs can avoid the hurdles posed by transplantation, a sustained treatment response with them will still require lifelong treatment.8 MSCs should still be considered for future CF therapeutic purposes, but new developments and techniques are required to optimize their efficacy.
Further advances in stem cell research have also made it possible to use somatic cells to generate patient-specific self-renewing pluripotent cells capable of directed differentiation into the specific epithelial cell types involved in the pathophysiology of CF.9–13 When used in combination with genome-editing technology, these techniques can yield new epithelial tissue with fully functional CFTR proteins.14,15 In theory, this disease-free tissue could then be transplanted back to the donor with minimal risk of transplant rejection. While the development of iPSC-based therapies for the treatment and potential cure of CF is promising, more research is needed to determine the safety and clinical feasibility of these techniques.
Conclusions
Novel gene-editing mechanisms, such as the use of CRISPR/Cas9 against the progenitor cells of the respiratory tract or novel lentiviral-mediated gene transfer, provide a window to a possible cure for CF. With further clinical trials, these gene therapies have the greatest potential to provide long-lasting CF treatment because of their ability to target the root genetic cause. Therefore, it is conceivable that, once the delivery mechanisms of genomic-editing molecules and the technical and safety barriers behind their usages are determined, they will lead the forefront of CF treatment alongside other stem cell–based methods.