Introduction
Thrombocytopenia is a decrease in platelet count to below the lower limit of normal, typically defined as 150,000/μL or 150x109/L platelets.1 This may occur secondary to suppression of platelet production or an increase in peripheral platelet destruction, clearance, or consumption. Commonly, platelets are found to be low in simple consumptive coagulopathy such as deep vein thrombosis or pulmonary embolism,2 as well as in disseminated intravascular coagulation.3 Drug-induced thrombocytopenia can occur by several mechanisms. Chong et al4 outline 3 mechanisms of drug-induced thrombocytopenia: (1) nonimmune, direct toxic effects; (2) an immune mechanism mediated by a drug-dependent antibody; and (3) autoimmune destruction mediated by a drug-independent antibody. Most drugs are thought to cause drug-induced thrombocytopenia by the second, drug-dependent immune-mediated mechanism.4
Drug-induced thrombocytopenia, specifically purpura related to quinine, was first described in 1865,5 and currently, more than 200 medications are implicated in causing it.4 Heparin-induced thrombocytopenia, the most common cause of drug-induced thrombocytopenia,6 has been documented for more than 70 years.7,8 However, to our knowledge, only 2 English-language reports have been published describing immune-mediated thrombocytopenia related to oral amiodarone,9,10 the first appearing in 1987.9 At least 8 reports detail nonimmune (direct toxic) effects of amiodarone on the bone marrow, resulting in pancytopenia.11–18
Amiodarone is grouped with the class III antiarrhythmic agents, but its mechanism of action is multifaceted. It causes peripheral and coronary artery vasodilation, as well as negative ionotropic, chronotropic, and dromotropic effects.19 These effects are mediated by the inhibition of potassium rectifier currents, voltage-gated sodium channels, β-adrenergic receptors, and calcium channels.20 Its efficacy has been documented in the management of rapid ventricular rate in the setting of both atrial fibrillation and advanced cardiac life support protocol in the setting of cardiac arrest.21–25 Widely described adverse effects include pulmonary toxicity24,26; thyroid dysfunction27; skin reactions, such as phototoxic erythema or hyperpigmentation28; neurologic effects, including tremor, ataxia, gait disturbance, and paresthesia29; ocular manifestations (in severe cases leading to permanent blindness)24,30; abnormal liver function test results24; and bradycardia.29,31
Case Presentation
An 89-year-old woman with a history of atrial fibrillation, hypertension, and coronary artery disease presented to the emergency department on request of her primary care physician after routine bloodwork revealed a platelet count of 8x109/L accompanied by a bilateral lower extremity petechial rash. On presentation, the patient had no acute pain, fever, chills, headache, cough, or visual changes, and she had not recently taken antibiotics. She denied epistaxis, hematuria, hematochezia, melena, and postmenopausal bleeding.
Her medical history was significant for ground-level fall with left temporal intraparenchymal and intraventricular hemorrhage about 2 months prior to her current hospitalization. During her 10-day hospitalization for this fall, the patient’s platelet counts were low-normal. The lowest recorded count was 124x109/L; at the time of her discharge to a subacute rehabilitation facility, her platelet count was 182x109/L. The patient’s most recent echocardiogram was performed during this hospitalization for intraventricular and intraparenchymal hemorrhage, and it did not show evidence of aortic stenosis. She started treatment with oral amiodarone, 200 mg, daily during this stay, which was 47 days prior to presentation with thrombocytopenia. The patient had continued with this dose until her presentation at our hospital.
On presentation, the patient’s vitals were within normal limits, including regular ventricular rate (81 beats per minute) and rhythm. Cardiac examination did not reveal murmurs, rubs, or gallops. Physical examination was remarkable for petechial rash on the patient’s bilateral lower extremities, but she did not exhibit respiratory distress, rales or wheezing, abdominal tenderness to palpation, hepatosplenomegaly, or lymphadenopathy. Furthermore, her neurologic examination was nonfocal.
Her laboratory workup was significant for a platelet count of 7x109/L (normal range, 150-400x109/L), red blood cell count of 3.59x106/µL (normal range, 4.2-5.4x106/µL), hemoglobin level of 10.7 g/dL (normal range, 12-16 g/dL), hematocrit level of 33.2% (normal range, 37%-47%), red blood cell distribution width at 16.4% (normal range, 11.5%-14.5%), total bilirubin level of 1.8 mg/dL (normal range, 0.2-1.3 mg/dL), blood urea nitrogen level of 26 mg/dL (normal range, 7-17 mg/dL), creatinine level of 1.07 mg/dL (normal range, 0.51-0.95 mg/dL), and glomerular filtration rate of 48 mL/min/1.73m2 (normal >60 mL/min/1.73m2). She also had a high granulocyte percentage (78%) and low lymphocyte count (13%), although her white blood cell count (7.60x103/µL; normal range, 4.8-10.8x103/µL) was normal. Prothrombin time, international normalized ratio, and partial thromboplastin time were normal at 13.2 seconds, 1.02, and 28.1 seconds, respectively. Her D-dimer level was elevated at 1.77 μg/mL (normal <0.90 µg/mL). Her fecal occult blood and hepatitis C test results were also negative.
Heparin-induced thrombocytopenia antibody was negative at 0.129 optical density (OD) (normal range <0.399 OD). Hemolytic processes were ruled out, with normal levels of lactate dehydrogenase at 208 U/L and haptoglobin at 115 mg/dL (normal ranges, 84-246 U/L and 30-200 mg/dL, respectively). Additionally, no platelet antibodies were detected, including antibodies to glycoprotein (GP) IIb/IIIa, Ia/IIa, Ib/IX, and IV. No antibodies to human leukocyte antigen class I antigens were detected. Although a blood smear was not available, the laboratory reported that the platelet count was “verified by microscopic examination.” Pertinent laboratory findings are summarized in the Table.
Head computed tomography did not reveal acute intracranial findings, and chest computed tomographic angiogram was negative for pulmonary embolism. Venous duplex of the bilateral lower extremities ruled out deep vein thrombosis.
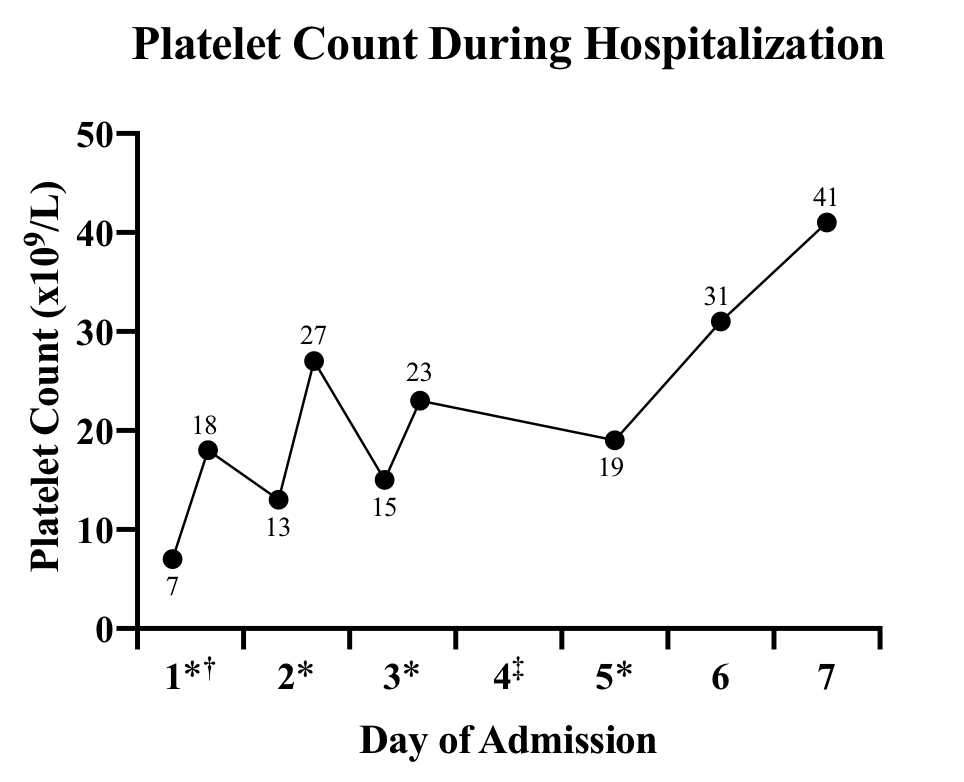
The patient’s other medications included aspirin, 81 mg, daily; atorvastatin, 40 mg, every evening; cholecalciferol, 1000 units, daily; vitamin B12, 100 mcg, 3 times per week; levothyroxine, 150 mg, daily; Lidoderm patches as needed; losartan, 100 mg, daily; metoprolol, 100 mg, 2 times daily; mirabegron extended release, 50 mg, daily; and tramadol as needed. There had been no recent medication changes besides the addition of amiodarone 47 days prior to admission. Treatment with aspirin was stopped, and she began treatment with methylprednisolone, 125 mg, intravenously every 8 hours; platelet concentrate was ordered for infusion. She was admitted to the intermediate-level care unit. The patient received a total of 4 platelet transfusions over her 7-day hospital course, and amiodarone treatment was discontinued after the first transfusion of platelets (Figure 1).
Treatment results following admission were as follows:
-
One hour after transfusion, the patient’s platelet count improved to 18x109/L, but downtrend to 13x109/L was noted 4 hours after transfusion.
-
She received another transfusion with appropriate response to 27x109/L on day 2.
-
On day 3, her platelet count was 15x109/L. The patient received another unit of platelets, with uptrend to 23x109/L.
-
On day 4, she started a 3-day course of intravenous immunoglobulin because her platelet count remained low despite transfusions and systemic steroids.
-
On day 5, her platelet level again declined (19x109/L), and the patient received a fourth and final unit of platelets.
-
On day 6, her platelet level had improved to 31x109/L.
-
The patient was discharged on day 7, with a platelet count of 41x109/L.
The patient was discharged with a regimen of prednisone, 40 mg, daily, but she did not strictly adhere to the recommended therapy and stopped taking prednisone on the fifth day after discharge. At her hematology follow-up 8 days after discharge (15 days after admission), her platelet count was 71x109/L. Seven months after admission (the next available platelet count), the patient’s platelet count was 169x109/L, which was within the normal range.

Discussion
Drug-induced thrombocytopenia is typically thought to be caused by an antibody that recognizes an epitope on normal platelets only in the presence of the sensitizing drug.32 Other less common mechanisms of drug-induced thrombocytopenia include hapten-dependent antibodies, drug-inducing conformational changes of platelet GPs, autoantibodies that react with platelets even in the absence of the drug, and immune complex formation (very rare).32 Limited investigation has been conducted into the mechanism of amiodarone-induced thrombocytopenia, but when it was first described, Weinberger et al9 noted a positive lymphocyte stimulation test in association with amiodarone-induced thrombocytopenia. This finding led to a hypothesis that the mechanism could be a cell-mediated, delayed hypersensitivity reaction.9 More recently, Sahud et al10 followed up on 3 patients with amiodarone-induced thrombocytopenia and found that these patients had antibodies specific for platelet GPs Ia/IIa or IIb/IIIa. These drug-dependent antibodies provide evidence of drug sensitivity, but are not always present in patients with drug reactions (as was observed with our patient); thus, a novel drug precipitant method of detecting these antibodies was devised in their study.10 Sahud et al10 have hypothesized that lipophilic amiodarone molecules can become incorporated into platelet membranes or into lipophilic domains of surface GPs, thus modifying these GPs in a way that makes them a target of GP Ia/IIa and GP IIb/IIIa antibodies. This process might also represent a mechanism by which other drugs may cause thrombocytopenia.
The mechanism of amiodarone-induced thrombocytopenia could be similar to the mechanism of thrombocytopenia sometimes observed after GP IIb/IIIa blockade in antiplatelet therapy because these antibodies could theoretically share a target.33 However, this mechanism remains poorly understood. Another GP implicated in drug reactions, GP V, has been ascribed as the target in quinidine-induced and gold-induced thrombocytopenia, although its association with amiodarone is unknown.34
In the setting of drug-induced immune thrombocytopenia, patients usually develop petechiae or other signs of drug-induced thrombocytopenia after using a new drug for about 1 week or sometimes much later if a drug is used intermittently over a longer period.32 Platelet counts typically begin to recover within the first 2 days of drug discontinuation, and complete recovery is seen by 1 week, even in the absence of steroid use.35 Indeed, although steroids are often given in the context of unexplained low platelet counts, they are only helpful if idiopathic thrombocytopenic purpura is the cause.32 According to Rondina et al,36 when the nadir of thrombocytopenia is more than 2 weeks after initiation of the offending pharmacologic therapy, a nonimmune-mediated drug-induced thrombocytopenia—the mechanism of which is nonselective myelosuppression and subsequent reduction in megakaryocytes—is needed. Case reports have reported recovery from amiodarone-induced thrombocytopenia 7 to 16 days after initiating steroid treatment,9,10 longer than is normally seen with other drugs (range, 2-7 days). Amiodarone is a lipophilic agent and is cleared from the body very slowly—the half-life of amiodarone is estimated at 58 days.37 Its active metabolite, desethylamiodarone, has an even longer half-life.38 Additionally, it is known that bioavailability of some drugs is increased in the elderly population and, due to changes in body composition, lipophilic drugs in particular can have an increased volume of distribution.39 These pharmacokinetics may account for the longer recovery time observed in these cases and the prolonged recovery observed in our patient.
In addition to severe thrombocytopenia, our patient also displayed mild normocytic anemia (low hematocrit level, hemoglobin level, and red blood cell count; normal mean corpuscular volume level; and elevated red blood cell distribution width). There are rare cases in the literature describing pancytopenia secondary to amiodarone-associated bone marrow granulomas, which typically develop over a period of 2 years and resolve over a course of several months.12,17,18 The possibility of our patient developing bone marrow granulomas over a few weeks seems unlikely, and her normal white blood cell count and only slightly low hemoglobin and hematocrit levels argue against bone marrow granulomas as well as nonimmune-mediated myelosuppression as previously postulated by Rondina et al.36 However, her presentation at 47 days after initiation of amiodarone is significantly longer than presentations previously reported for immune-induced thrombocytopenia: between 6 and 14 days after starting treatment with amiodarone.9,10 It is possible that the patient’s platelet count may have dropped earlier, before being brought to medical attention. She did report using lotion on her petechial rash for at least several days before seeing her physician, and she was not able to remember exactly when her petechial rash appeared. There is no other indication as to when her symptoms may have actually begun or why she may have experienced a delayed presentation of thrombocytopenia.
In terms of platelet recovery, the patient’s thrombocytopenia had not resolved by the second complete week of amiodarone discontinuation at the time of her follow-up with hematology (platelet count, 71x109/L). It is most likely that her counts had been delayed from returning to normal because of her older age (and changes in pharmacokinetics that come with aging) as well as the very long half-life and lipophilic nature of amiodarone.32 There is also the remote possibility, discussed here, that the patient’s thrombocytopenia was not antibody mediated, and instead was due to direct toxic effects of amiodarone, possibly causing bone marrow granulomas or nonselective myelosuppression. Her platelet count was eventually recorded within normal limits 7 months after admission (169x109/L), although the exact timing of her return to baseline is unknown.
Ultimately, all of this patient’s antibody study results were negative, which could indicate that she experienced direct drug-induced thrombocytopenia with accompanying mild anemia rather than immune-mediated thrombocytopenia. However, as Sahud et al10 found, not all patients with thrombocytopenic reactions in response to amiodarone harbored antibodies that could be detected by conventional methods. The patient’s slow platelet recovery after discontinuation of amiodarone is consistent with prior case reports and seems likely to be due to the pharmacokinetics of the drug. It remains difficult to delineate immune vs nonimmune mechanisms in this patient without further, specialized testing as described in Sahud et al.10 Bone marrow biopsy to rule out bone marrow granulomas or direct marrow toxicity was considered inappropriate, as the American Society of Hematology recommends against using bone marrow biopsy as a diagnostic tool for a typical presentation of what was presumed to be immune-mediated thrombocytopenia.40 Our patient continued to exhibit an appropriate response to treatment with amiodarone withdrawal, platelet transfusion, systemic steroids, and intravenous immunoglobulin. Thus, bone marrow biopsy was deferred to future evaluation if her condition worsened.
Before concluding that this patient’s thrombocytopenia was in fact due to amiodarone, other causes of thrombocytopenia, such as consumptive coagulopathy, acute blood loss, mechanical hemolysis, decreased blood cell production secondary to liver disease, and kidney failure were ruled out.41 The patient’s platelet recovery in the setting of continuation of all other medications effectively rules out any other medication causes of low platelet count.35 As drug-dependent, antiplatelet antibodies can persist for many years and an immune mechanism was thought to be likely, amiodarone was placed on the patient’s allergy list.35
Conclusion
This report highlights the need to be cognizant of the possibility of amiodarone-induced thrombocytopenia when beginning therapy and to surveil patients with complete blood count monitoring. This patient experienced a late-onset thrombocytopenia after beginning amiodarone, followed by a delayed recovery to normal platelet counts. As previous literature has reported, platelet recovery after amiodarone-induced thrombocytopenia is longer than observed with other drugs. Although this timeline of late onset and delayed recovery is more consistent with previous reports of amiodarone-mediated direct nonimmune toxicity or bone marrow granulomas, the patient’s normal white blood cell count and only mild anemia argue against a nonimmune cause of thrombocytopenia. Her antibody test results were negative, but previous studies have shown immune-mediated amiodarone-induced thrombocytopenia even in the absence of antibodies.10 Therefore, we believe this patient’s thrombocytopenia was immune-mediated. Her particularly drawn-out timeline of onset and recovery can be explained by physiologic changes that happen with normal aging, such as slower metabolic processing of pharmaceuticals and a larger volume of distribution of lipophilic drugs, such as amiodarone. Physicians starting amiodarone treatment for their patients should consider checking platelet counts by the end of the first 2 weeks of amiodarone treatment,9 and an unexpectedly low platelet count after initiation of amiodarone should prompt consideration of discontinuation and further monitoring.
Consent for the writing for this case report was provided by the patient.